
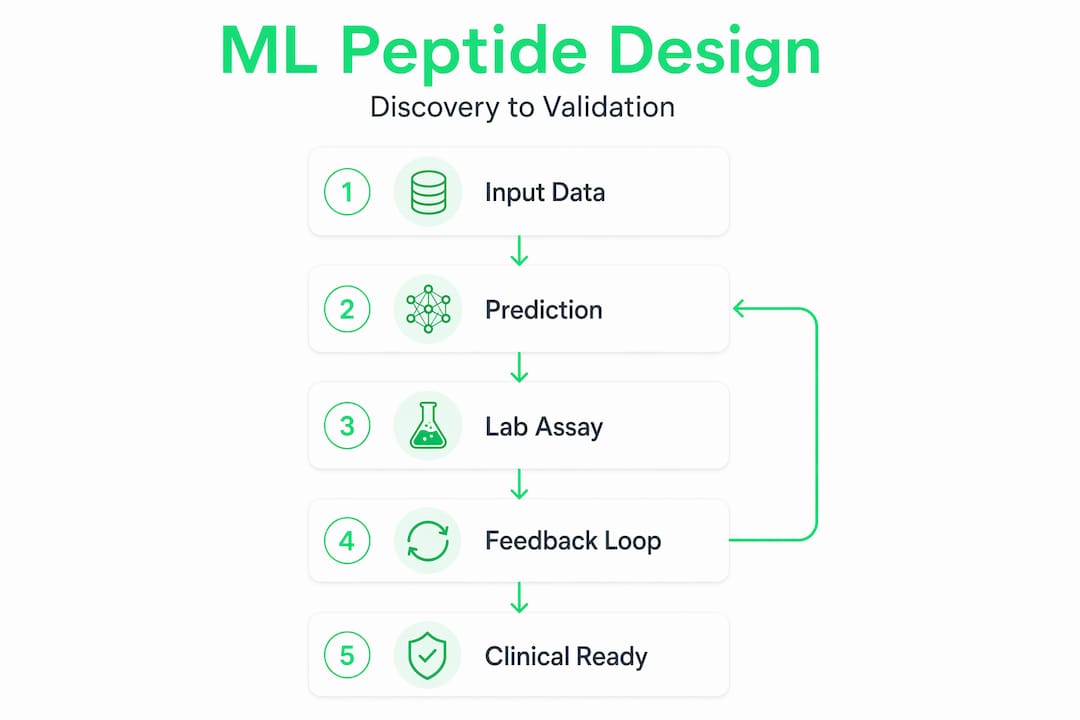
The old model of peptide discovery, where researchers screen thousands of compounds and hope for hits, is giving way to something far more precise. Machine learning peptides research has crossed a threshold in 2026: we now have experimentally validated closed-loop systems that adapt to real assay feedback, not just computational scores. Tools like ApexGO, CycloPepper, and TARSA are not theoretical. They are producing peptides that work in vivo, against drug-resistant bacteria and cancer cells alike. If you work in peptide therapy, research, or health optimization, the shift happening right now affects what you design, how you test it, and how fast you can get to a result that matters.
Table of Contents
- Key Takeaways
- Machine learning peptides: from one-shot prediction to closed-loop design
- Predicting synthesis success before you touch the bench
- Real-world applications in antimicrobial and anticancer therapies
- ML peptides in immunotherapy and precision health
- My honest take on where ML peptide design actually stands
- Track your peptide protocols with Peptideai
- FAQ
Key Takeaways
| Point | Details |
|---|---|
| Closed-loop ML outperforms one-shot models | Iterative feedback frameworks like ApexGO achieve an 85% hit rate, far exceeding traditional generative approaches. |
| Synthesis predictability is now computable | ML platforms predict cyclization success and aggregation risk before synthesis begins, saving time and resources. |
| Virtual screening at scale is practical | Reinforcement learning tools can screen 36 million peptide variants and reduce the viable search space by over 90%. |
| Immune receptor predictions are high accuracy | Protein language models predict KIR-peptide-HLA binding with AUROC above 0.8, opening new immunotherapy design pathways. |
| Multi-objective modeling matters most | Binding affinity alone is not sufficient. Manufacturability and toxicity must be co-optimized for clinical translation. |
Machine learning peptides: from one-shot prediction to closed-loop design
The most significant shift in machine learning in bioinformatics over the past two years is not a new algorithm. It is a new philosophy. Traditional generative models in peptide design were built to predict once and move on: train on known sequences, generate candidates, hand them off to the lab. The problem is that biological reality rarely matches the training distribution, and a single round of generation is rarely enough.
Closed-loop frameworks change this fundamentally. ApexGO integrates a transformer VAE with Bayesian optimization, continuously updating its generative model as experimental data comes back from the lab. The result is an 85% hit rate in antimicrobial peptide optimization, with in vivo efficacy confirmed. That number would be remarkable in any therapeutic modality.
The core architectural components driving this kind of performance include:
- Transformer-based variational autoencoders (VAEs): These encode peptide sequences into continuous latent spaces where interpolation and optimization become tractable, enabling exploration beyond cataloged databases.
- Bayesian optimization: Rather than random sampling, it models uncertainty explicitly and directs the generative model toward unexplored but promising regions of chemical space.
- Reinforcement learning: Used in platforms like TARSA, RL agents learn scoring functions from biological feedback, making them especially powerful for large-scale library searches.
- Protein language models (PLMs): Pre-trained on enormous protein sequence databases, PLMs bring transfer learning to peptide contexts where labeled data is scarce.
The exploration-exploitation tradeoff is where most ML peptide projects quietly fail. Bayesian optimization handles this by design. Treating peptide design as a latent-space optimization problem reduces wasted experimental iterations and surfaces better candidates earlier in the pipeline.
Pro Tip: When evaluating any AI for peptide design platform, check whether its optimization loop accepts real assay data as feedback or relies entirely on in silico scoring. The distinction separates tools that converge on real candidates from those that simply generate plausible sequences.
Predicting synthesis success before you touch the bench
One of the most underappreciated problems in peptide therapeutic development is not activity. It is making the peptide at all. Aggregation during synthesis, poor cyclization yields, and sequence-dependent degradation kill candidates that look excellent on paper. ML is now directly addressing this.

A 2026 Nature Chemistry study demonstrated that ML models can predict aggregation propensity during chemical synthesis, with the key finding that amino acid composition, not the exact sequence order, drives most of the aggregation risk. That insight has direct practical consequences: you can flag high-risk candidates before synthesis begins, simply by analyzing compositional features.
For cyclic peptides, the challenge is more specific. Cyclization site outcomes are difficult to predict empirically, and failed cyclizations waste significant synthesis effort. CycloPepper addresses this directly:
- ML training on 306 cyclic peptides produced a model that predicts head-to-tail cyclization outcomes with 84% prediction accuracy and 86% experimental consistency.
- Standardized data collection via CycloBot automated synthesis workflows ensure the training data reflects real synthesis conditions rather than inconsistent manual protocols.
- Synthesis feasibility scoring is embedded in the prediction pipeline, so researchers see manufacturability alongside predicted bioactivity.
The table below summarizes how these two synthesis prediction approaches compare:
| Platform | Target problem | Accuracy | Key input |
|---|---|---|---|
| CycloPepper | Cyclization site outcomes | 84% prediction, 86% experimental | Cyclic peptide sequence and cyclization sites |
| Nature Chemistry ML model | Aggregation during synthesis | Comparable across architectures | Amino acid composition encoding |
Multi-parameter ML models that capture developability alongside bioactivity are not optional for clinical translation. They are the difference between candidates that reach the clinic and those that stall in development.
Pro Tip: Prioritize ML workflows that integrate manufacturability scores from the start. A peptide that cannot be synthesized reliably at scale will never help a patient, regardless of its predicted binding affinity.
Real-world applications in antimicrobial and anticancer therapies
This is where the discussion moves from methodology to validated outcomes. Two platforms stand out in 2026 for producing experimentally confirmed results against serious disease targets.
ApexGO's closed-loop design framework has generated antimicrobial peptides with in vivo efficacy, confirmed against drug-resistant bacterial strains. The 85% hit rate is not a computational benchmark. It reflects candidates that passed wet-lab validation. For researchers working on antibiotic alternatives, this is the kind of performance that makes ML worth the investment in pipeline development.
TARSA takes a different approach, applying scalable reinforcement learning for anticancer peptide discovery. The numbers here are striking:
- Screened a virtual library of 36 million peptide variants using deep reinforcement learning and Markov Chain Monte Carlo sampling.
- Reduced the viable search space by 92.5%, identifying 15 cytotoxic candidates selective for resistant breast cancer cells.
- Validated selective non-toxicity, meaning the identified peptides did not harm healthy cells at therapeutic concentrations.
The implications for wet-lab workflow are significant. Ultra-large virtual screening via peptide classification algorithms compresses what would otherwise require years of empirical screening into a computation that can be completed in days. The wet-lab burden shifts from screening breadth to validation depth.
That said, limitations remain. Iterative experimental feedback is necessary to prevent ML models from converging on local optima that look good computationally but underperform biologically. No matter how sophisticated the model, the loop between prediction and experiment cannot be eliminated. It can only be made more efficient.
For health professionals tracking research-backed peptide lists in therapeutic contexts, the distinction between computationally proposed and experimentally validated candidates matters enormously.
ML peptides in immunotherapy and precision health
Predicting peptide interactions with immune receptors is one of the hardest problems in computational biology. The space of possible peptide sequences is vast, the structural data for many immune receptor complexes is limited, and allelic variation in HLA genes means that predictions must account for enormous biological diversity. Protein language models are now making meaningful progress here.
Protein language models predict KIR binding to peptide-HLA complexes with AUROC above 0.8. This is a clinically significant accuracy threshold for natural killer cell receptor interactions relevant to both viral infection defense and cancer immunotherapy. The model generalizes across allelic variants without requiring separate training for each HLA type, which is where deep learning protein analysis provides an advantage no traditional docking simulation can match.
The practical implications for immunotherapy peptide design include:
- Personalized neoantigen screening: ML enables rapid screening of tumor-specific peptides for individual patients, identifying those most likely to activate NK cells or T cells based on HLA profile.
- Viral defense applications: Predicting which peptides modulate KIR-HLA interactions has direct relevance to designing immunomodulatory peptides for chronic viral infections.
- Reduced experimental burden: High-accuracy in silico predictions allow researchers to prioritize the top 1% of candidates for synthesis rather than testing hundreds experimentally.
For anyone working on personalized peptide therapy, this immune receptor prediction capability represents a real near-term clinical tool, not a distant research aspiration.
My honest take on where ML peptide design actually stands
I've been watching the machine learning in bioinformatics space closely enough to say this plainly: the gap between what ML can predict and what researchers actually do with those predictions is still the biggest bottleneck in the field.
What I find genuinely exciting about 2026 is that the closed-loop frameworks have forced a more honest conversation about what optimization actually means. In my experience reviewing research and tracking platform development, the teams producing real results treat peptide design as an iterative control problem, not a one-time computational output. They run the loop. They let assay data reshape the model. They do not trust generation without validation.
What frustrates me is how many practitioners still treat ML as a black box that produces a shortlist. They skip manufacturability assessment entirely and are surprised when their top-ranked candidates fail synthesis. The research is clear: binding prediction alone is insufficient for clinical translation. You need multi-parameter models from day one.
The combination of automated synthesis platforms like CycloBot with ML prediction pipelines is the direction I think everything is heading. When the synthesis data feeds back into the model in real time, you close the loop that has historically required months of manual iteration. That is not a distant possibility. It is happening now, in published work, with validated results.
My advice to anyone building a peptide ML workflow in 2026: start with the synthesis constraints, not the activity predictions. If your candidate cannot be made reliably, the activity data is irrelevant.
— Sam
Track your peptide protocols with Peptideai
If the research covered here has you thinking seriously about your own peptide therapy approach, Peptideai is built for exactly this use case.
Peptideai brings research-grade intelligence into a mobile app that lets you build, track, and refine custom peptide stacks including BPC-157, TB-500, Semax, and 50 other cataloged peptides with precise dosing schedules. The AI Insights Chatbot delivers real-time, data-backed recommendations tied to your biometric data, and the AI Body Scanner tracks physical transformation over time. Seamless integration with Apple Health, Oura Ring, and Whoop means your wearable data informs your protocol, not just your intuition. Whether you are a researcher, a clinician, or a serious health optimizer, Peptideai puts validated peptide science in your hands. Explore what ML-informed peptide design looks like when it becomes a daily practice.
FAQ
What is machine learning's role in peptide design?
Machine learning accelerates peptide discovery by predicting bioactivity, synthesis feasibility, and receptor interactions before any synthesis occurs. Closed-loop frameworks like ApexGO further refine candidates by incorporating real experimental feedback into the optimization process.
How accurate are ML models for predicting peptide synthesis outcomes?
CycloPepper achieves 84% prediction accuracy for cyclization outcomes, while composition-based aggregation models from a 2026 Nature Chemistry study show comparable performance across multiple encoding architectures. Both significantly reduce synthesis failure rates.
Can ML replace wet-lab testing for peptide discovery?
No. ML reduces experimental burden by prioritizing candidates and flagging synthesis risks, but iterative experimental feedback remains necessary to validate and refine predictions. The most effective workflows treat ML and wet-lab work as complementary.
What are protein language models used for in peptide research?
Protein language models predict how peptides interact with immune receptors like KIR and HLA complexes, achieving AUROC above 0.8. They are especially useful for immunotherapy peptide design and personalized neoantigen screening.
How does ML improve antimicrobial peptide development?
ApexGO's generative optimization achieved an 85% hit rate for antimicrobial peptide candidates with confirmed in vivo efficacy, demonstrating that closed-loop ML substantially outperforms traditional one-shot generative approaches for antibiotic discovery.